PhD project: Effect of ’X’ on Aβ42 accumulations associated with Alzheimer’s disease
Visse Theresia Skov Moestrup, April 2016
Main supervisor: Arne Møller, Associate Professor
Alzheimer's disease
(AD) is the most common cause of dementia and is characterized by progressive neurodegeneration caused by cell shrinking, synapse loss, and neuronal death. One of the main contributors to these neuronal changes is accumulation of the protein beta amyloid (Aβ). Aβ not only accumulates in large extracellular plaques but also in the intracellular organelles and the plasma membrane. These accumulations are mostly caused by Aβ42. In the past 15 years, soluble Aβ42 has been shown to have deleterious effects on synaptic and cognitive functions. However, no clinical trials successfully decreasing Aβ42 production or enhancing extracellular Aβ42 clearance have so far been completed. Thus, there are no treatments to reverse or halt the progression of AD.
Our collaborator, Shanghai Advanced Research Institute, has recently shown that a compound (X) has dramatically reduced Aβ42 accumulations, and greatly suppressed the neural and behavioural deficits observed in a Drosophila AD model.
Mice models
In this PhD project, we firstly investigate the effects of X on Aβ42-deposits in the APP/PS1 mouse model of AD. The model has thoroughly documented characteristics such as increased hippocampal-Aβ42-load, decreased cerebrospinal fluid (CSF)-Aβ42-load, and memory impairments in the taste-aversion-test conducted in our laboratory (Figure 1; Ramirez et al. 2009).
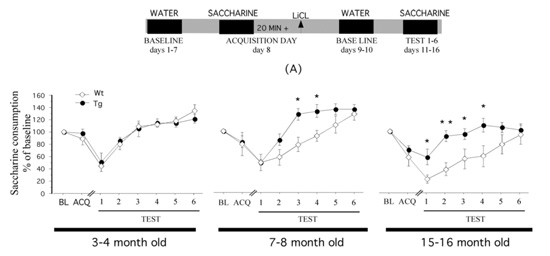
To investigate Aß-load in the APP/PS1 model, we will conduct immunohistochemistry of hippocampal brain-slices and ELISA quantification of CSF. To evaluate memory-impairments we will perform a taste-aversion behavioural test.
Organotypic cell cultures
Secondly, we will use organotypic cell cultures. We will introduce the cultures to Aβ protein and hypoxia, following treatment with X, and investigate the capability of X to decrease the amount of Aβ. Furthermore, we will investigate X in untreated cultures, to gain knowledge of the effect of X on healthy neurons.
PET imaging
Thirdly, we will investigate the capability of X to cross the blood brain barrier (BBB) in pigs. These investigations will be performed by the use of positron-emission-tomography (PET) imaging of radioactively labelled X.
Dog studies
Finally, we aim to investigate if the effects shown in the APP/PS1 model are transferable to aged dogs, suffering from canine cognitive dysfunction (CCD). CCD is, like AD, a neurodegenerative disorder characterised by elevated Aβ42-load and memory deficits (Figure 2; Fast et al. 2013). The effects of X in CCD dogs will be evaluated through behavioural tests, questionnaires for dog owners, and PET scans.
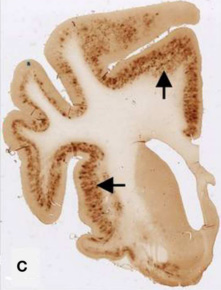
Figure 3: The brain of a 17-year old small mixed breed dog with clinical diagnosed CCD. The slice is immunostained with amyloid (Aβ) monoclonal 6E10 Dark areas identifies positive areas of immunostaining indicating deposits of Aβ, AβPP, or both.
Outcome
Through this project we expect to show a potent effect of X in: 1) mice models: hippocampus- Aβ42-load decrease, CSF-Aβ42-load increase, memory deficits improve, 2) organotypic cell cultures: to decrease the amount of Aβ, 3) non-rodents: BBB-permeability, and 4) CCD dogs: memory deficit improvement.
The present study will provide important information of the effects of X. Proof of the positive effects of X, this compound may be tested in clinical trials in humans and eventually be implemented in AD drug therapy.